
510(k) Expired
If the risk landscape evolves faster than the regulatory assumptions used to justify a clearance, what does it mean for a device to remain perpetually valid?
RISK MANAGEMENT
Manfred Maiers
3/13/20269 min read


510(k) Expired
In the United States, a medical device cleared through 510(k) or approved through PMA does not “expire” in any meaningful regulatory sense. The clearance or approval is not stamped with a renewal date. There is no automatic sunset. The device can remain commercially available indefinitely, as long as the manufacturer stays within the boundaries of what is considered the same device and continues to meet ongoing obligations like quality system compliance, complaint handling, and post-market reporting.
That permanence is not inherently irrational. It reflects a philosophy that the regulatory system should be stable, predictable, and anchored in change control. If the device has not changed in a way that alters its safety or effectiveness profile, why force a resubmission just because time has passed.
But the modern MedTech environment challenges the assumption embedded in that logic. The device may remain “the same” on paper while the world around it changes dramatically. Clinical workflows evolve. Hospital IT becomes a high-risk attack surface. Human factors expectations mature. Software becomes embedded everywhere. Cybersecurity moves from “nice to have” into a patient safety domain. Standards and consensus expectations shift repeatedly.
So, here’s the uncomfortable question that sits behind the title.
If the risk landscape evolves faster than the regulatory assumptions used to justify a clearance, what does it mean for a device to remain perpetually valid?
Regulatory architecture in one sentence
The U.S. system primarily treats clearance or approval as an event tied to a device and its change control, while the EU MDR transition treated regulatory adequacy more as a periodically revalidated state for access to the market.
That difference is not just bureaucratic. It shapes how legacy risk accumulates.
What “perpetual” means in practice in the U.S.
In the U.S., the core mechanism that prevents stagnation is supposed to be change control. Significant modifications that could affect safety or effectiveness should trigger regulatory interaction, potentially including a new submission. That approach assumes two things.
First, that meaningful risk changes are mostly driven by changes the manufacturer makes to the device. Second, that the manufacturer’s internal change control process is the right sensor for determining when regulatory reassessment is necessary.
Those assumptions are increasingly fragile.
Many risk shifts are environmental rather than design-driven. Threat models change. Use environments change. Interoperability demands change. Patient populations shift. Digital infrastructure becomes entangled with the device’s operation. None of that necessarily requires the manufacturer to change the device in a way that triggers a new submission, yet each can materially change real-world risk.
What the EU MDR transition signals conceptually
The EU MDR transition is often experienced by manufacturers as a forced modernization event. Even though the device itself did not change, the bar for demonstrating clinical evidence, post-market surveillance planning, and overall compliance readiness tightened. In other words, legacy status became less protective.
The practical outcome is that EU market access became more conditional on updated demonstration of conformity, not just historical placement on the market.
That is the essence of the contrast.
Comparison table: U.S. perpetual clearance vs EU MDR re-certification model
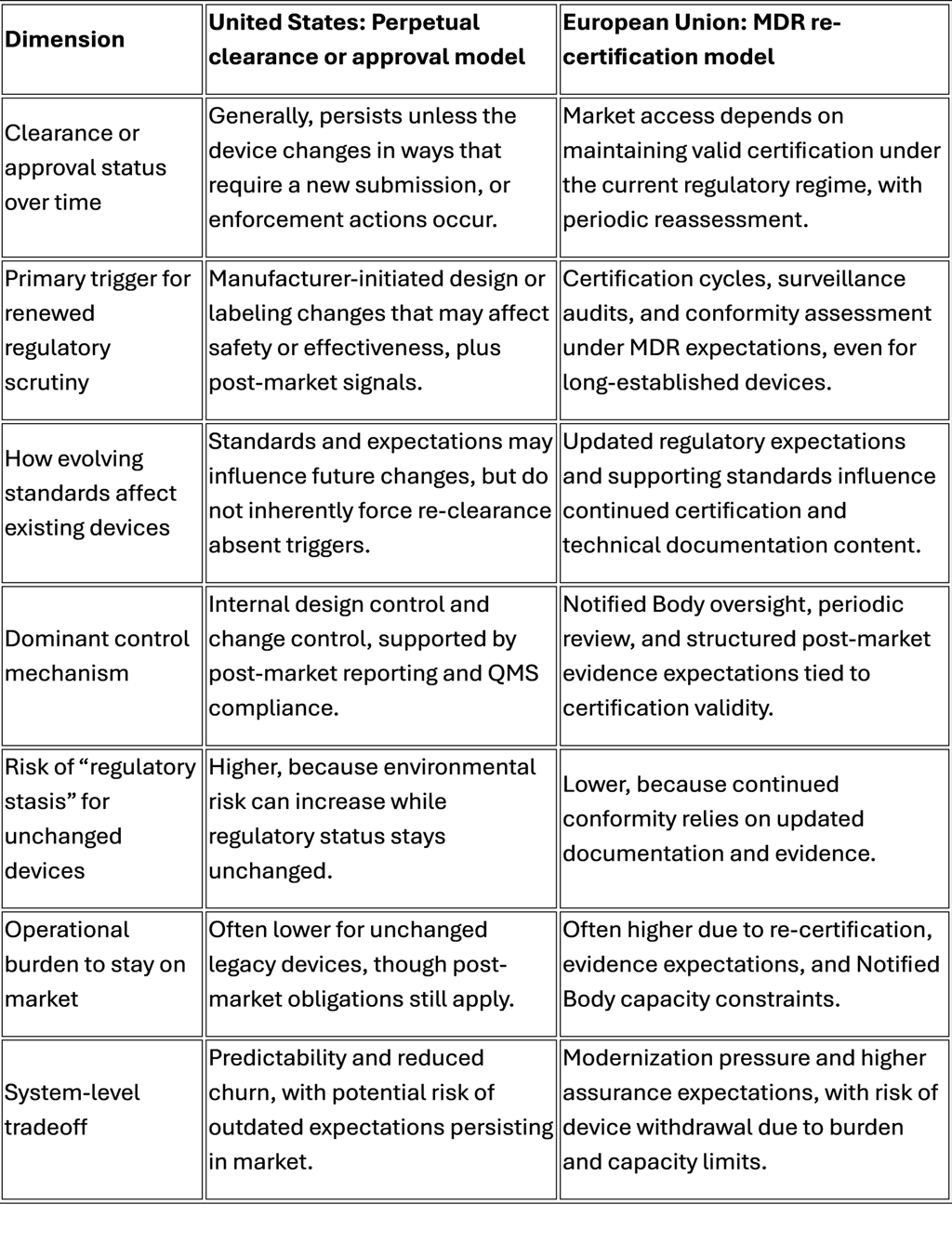
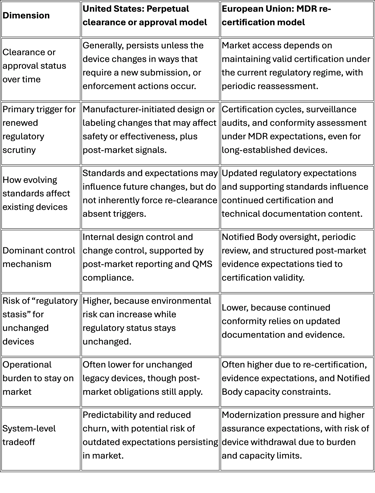
This table is not a moral scoreboard. It highlights a structural choice.
Perpetual status optimizes continuity. Re-certification optimizes refresh.
The question is whether the U.S. has enough “refresh energy” in its system when the world changes faster than design change control captures.
The legacy device problem is not age, it’s drift
“Legacy device” is often framed as “old device.” That framing is too shallow. The real issue is drift.
Drift is the gap between the assumptions that were reasonable at the time of clearance and the environment in which the device actually operates today.
Drift happens across multiple domains.
Cybersecurity drift
A device cleared in an era of isolated clinical equipment now lives in a networked hospital. That hospital has remote access pathways, vendor service connections, identity systems, patching constraints, and third-party integration layers. If the device was not designed with security controls aligned to modern threat models, it could become an entry point.
This is not abstract. Cybersecurity incidents can degrade availability, integrity, and confidentiality, and those can translate into patient harm when a therapy is delayed, a device is unavailable, or data integrity is compromised.
The core problem is that cybersecurity is not a one-time attribute. It is a moving target. A perpetual clearance model aligns poorly with moving targets unless it is paired with a robust lifecycle obligation that forces continuous improvement.
If the improvement is optional, drift grows.
Human factors drift
Human factors is another domain where expectations have matured. Devices designed when usability engineering was a softer discipline may not handle modern workflow realities well. Alarm fatigue, UI ambiguity, confusing affordances, and inconsistent interaction patterns can all drive use error.
The crucial point is that these issues are not always “defects” in the traditional sense. They are mismatches between design and context. Context evolves.
Perpetual clearance can allow devices to persist that are operationally “acceptable” but misaligned with modern safety culture and usability expectations.
Software and connectivity drift
A surprising number of devices that started as electro-mechanical products have accumulated software dependence over time. Even if the core therapy mechanism is unchanged, the surrounding ecosystem changes.
Operating systems become obsolete.
Drivers and interfaces are no longer supported.
Integration expectations rise.
Telemetry becomes standard.
Data becomes part of clinical decision-making.
If a device is functionally safe but digitally brittle, that brittleness becomes a safety concern in a modern system.
Standards drift and the illusion of equivalence
Consensus standards are one way industry encodes “what good looks like right now.” Standards evolve because problems are discovered, capabilities advance, and failure modes become better understood.
When standards change substantially, a device can remain legally marketable yet increasingly misaligned with contemporary definitions of adequate risk control. Over time, “substantially equivalent” can become a historical artifact rather than a meaningful safety proxy.
That is the “510 Expired” concept.
Not that the letter of clearance expires, but that the assumptions can.
Patient risk implications: where drift becomes harm
It is easy to talk about drift as an engineering philosophy problem. The stakes are higher.
The risk pathways to the patient generally cluster into a few families.
Safety risk via cybersecurity and availability
A compromised or unavailable device is not just an IT incident. In clinical care, availability is a safety function. If an infusion system cannot be used, if a monitoring device loses integrity, if configuration is altered, the downstream impact can include delayed therapy, wrong therapy, or degraded situational awareness.
Even without malicious intent, lack of patchability or insecure remote support pathways can create chronic exposure that hospitals are forced to accept for operational reasons.
This shifts risk from manufacturer design space to hospital mitigation space. Hospitals are not always equipped to compensate.
Safety risk via usability and workflow mismatch
Use error is rarely the result of “bad users.” It is usually a predictable outcome of design and context. As clinical staffing models, workload, and training patterns evolve, devices with high cognitive load and ambiguous UI design become more dangerous.
A legacy device can therefore create a persistent safety tax on the clinical system.
That safety tax shows up as.
Additional training burden.
Greater reliance on workarounds.
Higher chance of error during high-stress operation.
Increased alarm burden.
If you want practical framing, ask: does this device require a hero to operate safely?
If yes, the system is paying for the device with human risk.
Safety risk via maintenance, obsolescence, and parts substitution
Legacy devices often live long enough to encounter part obsolescence. Parts get substituted. Service methods evolve. Third-party servicing emerges. Refurbishment becomes common. Each of these can be safe, but each also increases variability.
Variability is the enemy of assurance.
When a device’s manufacturing baseline is decades old, maintaining consistent performance and consistent risk controls becomes harder. The device can stay on the market while the actual instances used in hospitals become increasingly heterogeneous.
From a risk management perspective, you begin to lose confidence that “the device” is a single coherent entity.
Safety risk via outdated clinical assumptions
Some devices are cleared based on intended use and clinical practice patterns that have since changed. Even if the labeled intended use is stable, real-world use patterns may drift. Off-label use may become normalized. Patient populations may shift.
A system that never forces periodic re-justification risks enabling clinical mismatch at scale.
The hospital and physician reality: installed base is its own regulator
Even if the market stopped selling a legacy device tomorrow, hospitals can keep using installed equipment for years. That installed base creates a second arena of risk: operational persistence.
Hospitals optimize for.
Capital expenditure constraints.
Maintenance continuity.
Interoperability constraints.
Standardization across units.
Training and staffing stability.
These are rational constraints, but they create a situation where the device lifecycle is driven as much by hospital economics as by patient risk.
When a legacy device has known cybersecurity weaknesses, the hospital may respond with segmentation, compensating controls, and operational constraints. Those mitigations are not free. They add complexity and failure modes of their own.
When a legacy device has usability limitations, the hospital may respond with training and procedural controls. Those controls are also not free. They rely on human compliance.
In both cases, risk becomes distributed across the care system rather than engineered out at the product level.
This is the hidden patient safety cost of perpetual clearance: it externalizes modernization responsibility.
What options exist besides doing nothing
A simple slogan like “force a new 510(k) whenever standards change” sounds clean. In practice, it is complicated. Standards change often. Not all standards matter equally. Not all devices share the same risk profile. Regulatory bandwidth is finite. Industry capacity is finite. Hospitals cannot tolerate device shortages.
So, the right question is not “should we force resubmission.”
The right question is “what mechanism creates a predictable, risk-based refresh cycle without collapsing continuity.”
Here are several structural options worth serious consideration.
Option 1: Standards-triggered reassessment, but risk-tiered
Instead of tying reassessment to any standards change, define a tiered trigger model.
Tier A: Devices with significant cybersecurity exposure, software dependence, or high-severity harm potential.
Tier B: Devices with moderate harm potential and limited connectivity.
Tier C: Low-risk devices with minimal complexity.
When critical standards or regulatory expectations change in a way that affects essential safety domains, Tier A devices could require a structured reassessment package, not necessarily a full resubmission, but a formal demonstration of gap closure or justified equivalence under modern expectations.
This is the key design goal: avoid a binary “resubmit or not” gate. Create intermediate compliance artifacts that force modernization where it matters.
Option 2: Sunset for certain claims, not for marketability
A device might remain marketable, but certain claims or intended-use expansions could sunset unless refreshed.
For example, connectivity-related claims, interoperability claims, remote management claims, or software-driven performance claims could require periodic refresh. That targets drift where drift is most dangerous, while preserving continuity for core therapy mechanisms that have stable safety profiles.
This is analogous to narrowing the scope of “perpetualness.”
Option 3: Lifecycle cybersecurity and usability obligations with auditable outcomes
If the core problem is moving targets, then the remedy is lifecycle obligations. In practice, which means.
Periodic cybersecurity risk assessment updates tied to current threat models.
Coordinated vulnerability disclosure readiness and response expectations.
Patchability strategies and compensating control guidance.
Usability surveillance and systematic use-error learning loops.
To matter, these obligations need measurable outcomes and auditability. Otherwise, they become paperwork.
This approach is attractive because it does not require reopening the original clearance logic every time standards shift. It instead imposes a living safety posture.
Option 4: Installed-base modernization programs
Hospitals bear installed base risk. A policy approach could recognize that reality and incentivize modernization pathways.
Examples include.
Incentives for upgrades that remove high-risk cybersecurity exposure.
Procurement standards that require vendors to provide patch support for a defined period.
Shared responsibility frameworks that clarify who owns which mitigation layers.
The goal is to reduce the “forever device” phenomenon in practice, even if clearance stays perpetual.
Option 5: New submission triggered by “regulatory delta” thresholds
My original idea is the most direct: require a new 510(k) when standards or regulations have changed materially since the original submission.
The problem is defining “substantially.”
A workable implementation would need.
A published list of “delta thresholds” that are narrow and high-impact.
Clear mapping to device categories and risk profiles.
Transition timelines that prevent supply shocks.
Scaled evidence expectations that do not replicate full new-device burden.
If it is too broad, it collapses the system. If it is too narrow, it becomes symbolic.
The principle is still sound: when the basis of safety assurance changes meaningfully, the assurance should be refreshed.
The uncomfortable trade space
Every reform option introduces a tension between.
Patient safety modernization.
Supply continuity and device availability.
Regulatory capacity and predictability.
Innovation incentives and economic feasibility.
The EU MDR experience highlights one risk of aggressive refresh pressure: device withdrawal. The U.S. experience highlights one risk of low refresh pressure: drift and externalized mitigation.
The answer is not to copy one system. The answer is to correct the failure mode.
For the U.S., the failure mode is that perpetual clearance can behave like perpetual adequacy.
Those are not the same thing.
Conclusion: what “510 Expired” should mean
“510 Expired” is not an argument that every legacy device is unsafe. Many legacy devices are robust, clinically validated by decades of use, and operationally reliable.
It is an argument that a perpetual administrative status can mask a non-perpetual safety reality.
The core patient safety risk is not the age of the device. It is the ungoverned drift between old assumptions and new environments.
If we accept that drift is inevitable, then we need a mechanism that periodically forces one of two outcomes.
Modernize the device, its controls, and its lifecycle obligations to match contemporary risk.
Justify, with evidence, why modernization is not necessary for safety and effectiveness in today’s context.
Either result is better than silent drift.
The U.S. does not need to abandon stability to achieve modernization. It needs a deliberate refresh architecture that targets high-impact drift domains, especially cybersecurity, usability, and software-driven performance, and that acknowledges the installed-base reality in hospitals.
Perpetual clearance can remain a feature.
Perpetual complacency cannot.